Product Focus
The transition-metal-catalyzed cross-coupling reactions are powerful tools for the formation of carbon–carbon bonds. Among these transformations, the Suzuki–Miyaura reaction, which is the transition-metal-catalyzed cross-coupling between an organo-boron compound and an organic (pseudo)halide, has become the most attractive method since its discovery in 1979. While many organo-boron compounds have been discovered, N-methyliminodiacetic acid MIDA boronates represent tremendously useful building blocks for the Suzuki–Miyaura cross-coupling that has been successfully applied to sequential synthesis of various natural product motifs.
Organic azides are an important class of synthetic building blocks. In organic chemistry azides are commonly used for introducing an amino-group via Staudinger reaction. With the recent advent of "click-chemistry" azides became enormously popular for their participation in the Cu(I)-catalyzed Huisgen azide-alkyne 1,3-dipolar cycloaddition reaction. Because of the high rate of synthetic success of both Huisgen cycloaddition and Staudinger reduction these reactions are often used in combinatorial and medicinal chemistry. Enamine presented the set of azides suitable for click-chemistry transformations earlier.
Fragment-baseajord drug design has become a novel paradigm for drug discovery in the last decade. The lack of molecular rigidity intrinsic to a majority of small organic molecules appears to be a serious obstacle for this promising approach. Conformationally restricted rigid molecules show higher reproducibility of results when used in projects utilizing in silico screening methods. Due to a predefined spatial orientation of the molecular fragments mounted on a conformationally rigid scaffold, a decrease in entropy of molecular binding with its biological target might be expected, thus leading to higher affinity of the potential drug candidate.
This issue of Enamine Product Focus highlights Lactams, cyclic amide building blocks. There are numerous examples of Lactams usage in drug discovery, e.g., β-lactam based antibiotics, oral anticoagulant Rivaroxaban, and anticonvulsant Levetiracetam.
Rivaroxaban, 2008
Levetiracetam, 2000
The intrinsic role of heterocyclic compounds in medicinal chemistry is now undoubtful as more than 90% of new drugs contain heterocycles. Compounds possessing heterocyclic cores have several advantages that make their use in drug design particulary effective.
- Aromatic rings are extremely rigid in conformation thus defining spatial orientation of attached functional groups and reducing entropy of binding of the molecule with potential biological target.
- In most cases, hetero atoms exhibit properties of strong hydrogen bond donors/acceptors enforcing interaction of the molecule with its biological target.
- The chemistry of heterocyclic compounds is undoubtedly the most explored area of organic chemistry. The synthetic methods for the construction of heterocycles are well-established, simple, highly efficient and easy-scalable.
- Heterocycles occupy an overwhelming majority of the available chemical space thus being the most diverse class of organic compounds. Therefore they represent the substantial possibilities for the design of novel scaffolds that are yet not protected by patents.
Bifunctional building blocks are of special interest for drug design and organic synthesis due to three reasons, at least. First, these compounds can be used to tether two molecular fragments responsible for binding to the biological target, thus they can act as linkers. Second, if one functional group is not engaged in connection between the core of building block and the rest of a molecule being constructed, then it can participate in important interaction with a biological target. Finally, many bifunctional building blocks can undergo cyclization reactions, allowing rapid advance toward prospective heterocyclic units.
This issue of Enamine Product Focus represents a family of Building Blocks possessing sulfone moiety as a part of a cyclic unit. All the building blocks processing cyclic sulfone moiety have some common features that make the design of potential drug candidates particularly efficient.
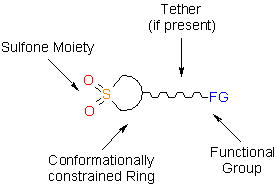
A concept of bioisosterism has attracted much attention in recent years. The success of this strategy in developing new substances which are therapeutically attractive has observed a significant growth in distinct therapeutic classes, being amply used to discover new analogs of commercially attractive therapeutic innovations, and also as a tool useful in the molecular modification.
A lead compound with a desired pharmacological activity may have undesirable characteristics that limit its bioavailability, or structural features which adversely influence its metabolism and excretion from the body. Bioisosterism represents one of the approaches used by the medicinal chemists for rational modification of lead compounds into safer and more clinically effective agents.
This issue of Enamine Product Focus represents a family of Building Blocks containing Hydrazine unit. Several aspects of utilizing the hydrazine building blocks for the design of potential drug candidates can be outlined. Despite hydrazines themselves are rarely thought as promising drug candidates, some successful examples of drugs possessing hydrazine moiety can be found, e. g. antiparkinsonic agent Carbidopa (Lodosyn®), antidepressant Phenelzine (Nardil®) or vasodilator Hydralazine (Apresoline®).
The synthesis of functional organic compounds for different applications largely relies on laborious approaches involving an extensive usage of protecting groups. Nowadays chemoselective routes to the construction of complex molecules are becoming a good alternative because they shorten the length of multistep syntheses by excluding protecting groups and, as a consequence, lead to increased overall yields.
Substituted ureas are drawing considerable attention as pharmacologically relevant compounds. Our proprietary trifluorourethanes, reported earlier, are suitable precursors for the preparation of aryl/heteroaryl-alkyl or aryl/heteroaryl-dialkyl urea derivatives via parallel synthesis, but they are not appropriate for the preparation of ureas bearing different alkyl substituents. We report now a new set of building blocks, carbamoylimidazoles, specifically designed for the synthesis of ureas with multiple alkyl groups.
The number of commercial pharmaceutical compounds which contain at least one fluorine substituent noticeably increased since 1950th, when the first fluorine-containing drugs were introduced. At present, about 20% of the commercial drug substances are fluorine-substituted organic compounds. As the prominent examples, top-selling anti-depressant Fluoxetine (Prozac), the cholesterol-lowering drug Atorvastatin (Lipitor), the antibacterial Ciprofloxacin (Ciprobay) could be named.
In the previous issue, we presented the set of building blocks which had at least one fluorine substituent. This issue continues representation of fluorine-substituted building blocks emphasizing compounds derived from original scaffolds designed by Enamine chemists.
Understanding the importance of the fluoro-organic compounds for the pharmaceutical industry, we put much effort in adapting known and developing new synthetic methodologies allowing introducing fluorine by the use of different fluorine reagents.
The primary structure of a peptide can be formally considered as a oligoethylenediamine molecular platform to which the side chains and carbonyl oxygen atoms are attached through single and double bonds respectively. This backbone is conformationally restrained at the amide bonds and has some rotational degrees of freedom at single C-C and C-N bonds. The later become restricted upon the formation of the secondary structures stabilized by multiple hydrogen bonds. The fragments of 1,3-, 1,4-, 1,5- and 1,6-diamines are found in peptides containing aspargine, glutamine, tryptophane, histidine, lysine and arginine. Apparently some structural, conformational and functional features of peptides can be reproduced by designed derivatives of synthetic conformationally restrained diamines.
This issue of Enamine Product Focus introduces a family of functionalized building blocks called linkers. The term “linker” has been used in medicinal chemistry to define different substances. To avoid ambiguity we will use here the following meaning: a compound is viewed as a linker if it has two or more chemically orthogonal functionalities on an inert, flexible or rigid scaffold. Functional groups could be used to connect (or “link”) fragments responsible for the interaction with biological target – this is applicable both for HTS libraries design and in Fragment-Based Drug Discovery (FBDD). For the latter approach to drug discovery, the design of linkers is particularly critical.
Amino acids are the building blocks which are widespread in the Nature. Even of the limited number of the genetically-coded α-amino acids the Nature composes practically unlimited number of proteins. The “success” of proteins in sustaining life and regulating biochemical processes prompted chemists over the world to mimic their structure and function in search for new drug candidates – peptidomimetics. It is therefore not surprising that the selection of the natural proteinogenic amino acids was substantially enriched by creating new, unnatural amino acids, specially designed to improve pharmacokinetic and pharmacodynamic properties of the peptidomimetics and other biologically active compounds based on them.
"The α-amino acids, molecules of which have restricted conformational flexibility, are widely used in design of peptidomimetics, peptide models, and in systematic search for biologically active compounds. Among these amino acids, a distinct class of compounds can be highlighted, namely - conformationally rigid amino acids (CRA). Certain torsion angles, which describe the conformation of a polypeptide chain at the CRA, are "fixed" that allows predicting and controlling it to some extent. Many structural studies show that the CRA residues can dictate certain conformation of the peptide chain around them, consequently, they can stabilize or destabilize certain peptide secondary structure elements.
For Palladium-Catalysed C–N And C–C Cross-Couplings.
Air and moisture insensitive auxiliary ligands for highly efficient palladium catalysts. These ultra-low loading (high TONs or TOFs) catalysts have shown excellent activity in Suzuki cross-coupling with aryl bromides and chlorides, Heck vinylation, Heck-Sonogashira alkynylation and allylic amination of allyl acetates (Tsuji–Trost type reactions). Low loadings of the catalysts ensure minimal contamination of the final compounds with palladium and simplify the purification procedures. High stability of the allows to store them for unlimited time without special precautions.
Recently 3-amino-tetrahydrothiophen-1,1-dioxides have drawn significant attention of a major drug developers. A few publications have been released earlier, corroborating rather high potential of this pharmacophoric moiety.
In the last decade, development of new drugs increasingly requires the use of chiral building blocks for hit-to-lead optimization, and even on early stages - in search for efficient hit compounds. The main fundamental reason for this lies in the fact that almost all the biological targets are chiral, and the drug-receptor interaction requires strict match of chirality. The formal reason results from strengthening the regulatory guidance for submitting new drug applications in Europe and USA which concern chirality issues.