Designed using generative AI and predictive models for the most efficient hit-finding
Enamine's REAL Space represents the world’s largest, continuously expanding make-on-demand virtual library, built on insights from millions of parallel syntheses. While the vast chemical space offers incredible potential, selecting the most promising compounds for successful drug development from billions of virtual molecules can be a daunting task. That's where AI and Machine Learning come in, enabling the creation of smaller, highly focused libraries that consistently outperform traditional large HTS collections. By integrating one of Recursion's cutting-edge AI/ML tools, MatchMaker, with REAL Space, we've curated 10 powerful screening libraries from 15 000 newly synthesized compounds, specifically designed to accelerate drug discovery with precision and speed.
Recursion (NASDAQ: RXRX), is a leading clinical-stage TechBio company, leverages sophisticated machine learning to decode biology with one of the world’s largest proprietary biological and chemical datasets, enabling precision and speed in drug discovery. Recursion’s tool, MatchMaker, uses machine learning to predict small molecule compatibility with multiple protein targets, offering a scalable, less computationally intensive alternative to traditional methods. This enables faster decision-making, enhances Recursion's datasets, and pre-screens for precision modeling to accelerate drug discovery.
What Makes this Chemical Compound Library Unique and Valuable?
Most commercial compound libraries cover broad sectors of biology such as GPCRs, kinases, or antibacterials that may include hundreds to thousands of individual proteins. Here we provide targeted libraries, subsets from the Enamine REAL Space, which are designed around common features seen when grouping proteins by their binding properties. These AI-enabled libraries were built around the hundred most promising and clinically relevant drug targets grouped into 10 families.
Development Strategy
As a first step, the human proteome was encoded into binding likelihood vectors through MatchMaker, a neural network model trained on a vast collection of biochemical datasets, to predict interacting protein-ligand pairs, otherwise known as Drug-Target Interactions (DTI). The entire Enamine REAL Space library was then scanned for putative interactions within the proteome, and the resulting pairwise matrix (compound vs protein) of 2.8 quadrillion interactions was aggregated at the synthon level. The emerging space is rich in chemical and biological information. It can be navigated to identify chemo-proteomic clusters: small, focused target sets predicted to bind with structurally and chemically similar compounds.
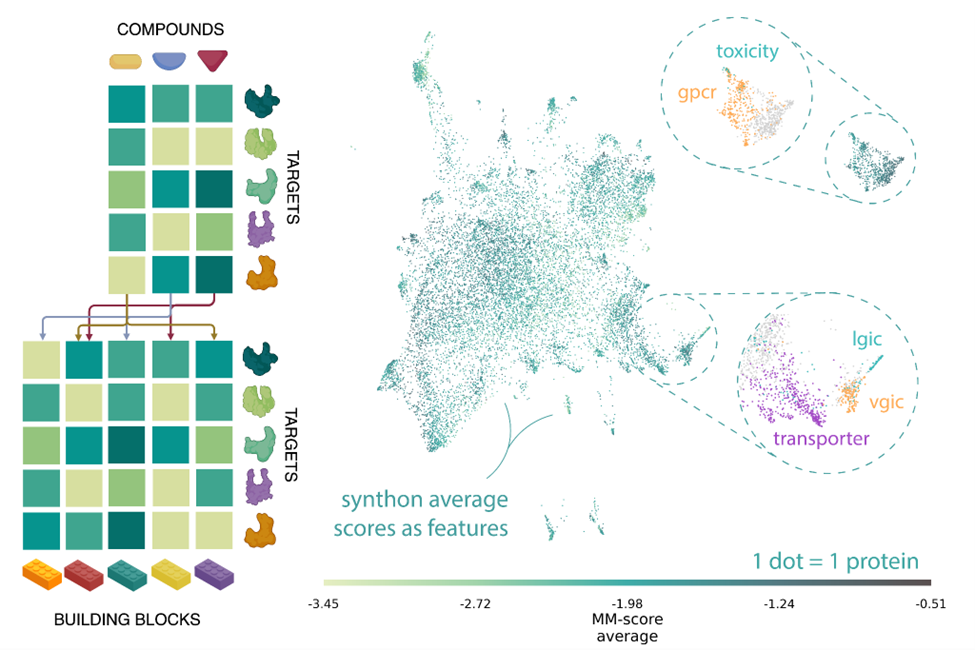
Left) MatchMaker ligand-target interaction likelihood scores were mapped onto a reduced-dimensionality space defined by synthons, theoretical building blocks of chemistry. Right) 2D projection of the human proteome as a function of their MatchMaker DTI prediction profile in synthon space. The feature space carries inherent biological and functional information as similar protein families are clustered together.
- Targeted libraries are designed starting from a list of seed targets relevant to biological pathways of interest and expanded to relevant chemo-proteomic clusters.
- Compounds were ranked against targets in each cluster according to their MatchMaker DTI prediction score.
- The final library set was selected to maximize diversity in drug-like, synthesizable chemical space, through the application of a range of classical medicinal chemistry, DMSO stability and synthesizability filters.
- 15 000 new compounds were synthesized to deliver highly focused AI-driven libraries
Libraries Catalog
Size
1 600
compounds
Description
Focused on targeting the top CRLs that have been explored as the most promising targets
Download file
Size
1 520
compounds
Description
Designed to target 3 HECT E3 Ligases for the discovery of new-generation drugs
Download file
Size
1 520
compounds
Description
Designed for discovery of RNF216, RNF19A, PRKN, RNF13 potent binders and modulators
Download file
AI-enabled USP Library
Size
1 200
compounds
Description
Designed for modulation of the largest DUBs family and delivery of a promising treatment for incurable diseases
Download file
AI-enabled GPCR Library
Size
1 760
compounds
Description
Designed to deliver new and efficient modulators of CCR5, HTR2A, HTR2B, MRGPRX1, CXCR6, CMKLR2, CCR10, GPR3, GPR4, GPR39, CCR4 receptors
Download file
HIPPO Pathway Library
Size
1 600
compounds
Description
Designed to deliver reliable hits for over 10 essential targets involved in the Hippo pathway
Download file
AI-enabled Molecular Chaperones Library
Size
1 360
compounds
Description
Designed for hit finding for Hsp90, Hsp70, Hsp60, HspD1, ClpP, Ch60 and Hsp100 protein targets
Download file
AI-enabled Allosteric Ion Channel Library
Size
2 080
compounds
Description
A new approach for modulating the most investigated and effective drug targets with good clinical records
Download file
AI-enabled PARP Library
Size
1 440
compounds
Description
Designed to target the most investigated and validated for drug discovery PARPs
Download file
AI-enabled TF Library
Size
1 520
compounds
Description
The hottest protein targets for discovering new treatments for the most challenging and not yet treated diseases
Download file
Support
We offer comprehensive support in developing your hit compounds. Naturally such programs are realised most efficiently when biological actives originate from our screening collection. However, even if the hit compounds are from the collections of other vendors lead identification and optimization projects can proceed most productively in our hands. Sometimes for this we only need to synthesize first examples of the given chemical series and validate synthesis route.