Accelerate drug discovery with precision and insight
Unlock the full potential of in silico drug discovery with Enamine’s advanced Computer-Aided Drug Design (CADD) services.
By partnering with us, you gain access to custom-tailored modeling workflows that integrate state-of-the-art molecular modeling, AI-powered screening, and medicinal chemistry expertise — all focused on maximizing your success rate in early-stage drug discovery.
Our team will support you through every step — from target identification and structure prediction, to hit identification, lead optimization, and beyond. We combine tools such as structure-based and ligand-based virtual screening, molecular docking, molecular dynamics, and free energy perturbation (FEP) to help you prioritize only the most promising candidates for synthesis and biological testing.
You don’t just get access to computational tools — you get a results-oriented strategy built around binding affinity prediction, ADMET profiling, and real-time decision-making, designed to reduce cost and timeline in your pipeline.
By collaborating with Enamine’s CADD department, you benefit from:- Deeper insight into target behavior and binding pocket dynamics
- Rapid identification and ranking of hits from Enamine REAL and screening libraries
- Integration of AI, ML, and cheminformatics to explore vast chemical space
- Seamless transition from virtual screening to synthesis and biological validation
Whether you're working with a challenging protein, RNA structure, or novel chemical series, our computational chemistry expertise helps you design, predict, and optimize with confidence.
We use a balanced approach employing different methods complementing each other
Target Modeling and Investigation
Homology Modeling
- 3D-structure prediction for unresolved targets
- Structure repairing
- Mutant modelling
Target Structure Analysis
- Potential binding site searching
- Active residue estimations
- AI binding site prediction
DNA/RNA - Complex Modeling
- Intercalation and binding in RNA groves
- G-quadruplex modeling
- Ryboswitches
- DNA GG and GA binding pocket
Molecular Dynamics (MD) Simulation
- Comparison and demonstration of the behavior of structures over time with and without a ligand
- Modeling the impact of mutations on stability, activity, and resistance to known binders
- Estimation protein-ligand binding energy
- Cryptic pocket searching
Ligand-based Drug Design (LBDD)
Analogs Similarity/Substructure Searching
- 2D searching (Morgan Fingerprints, ECFPs, etc.)
- 3D searching (QuickShape)
- Diversification/clustering
- Bemis-Murcko scaffolds/graph search
- Synthons search
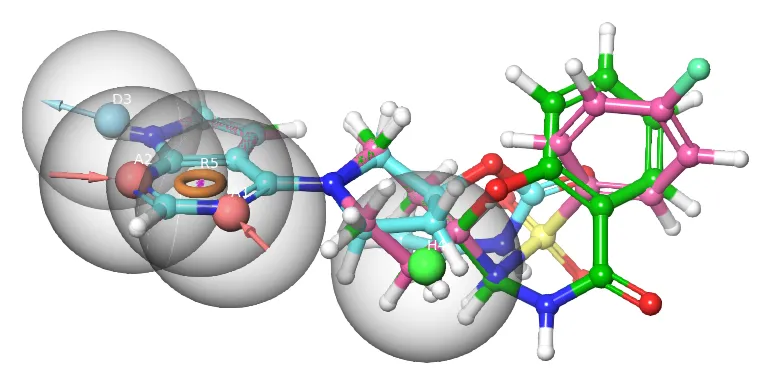
LB 3D pharmacophore virtual screening
ML Modeling and Prediction
- ML-powered high-throughput virtual screening
- ML-facillitated building of QSAR models
- ML-driven ADMET properties prediction
Hit-to-Lead Integrated Projects Support
- Evaluation of synthetic feasibility and screening result analysis
- Structure-activity relationship (SAR)
- Scaffold hopping/morphing
- Bioisosteric replacement
Electronic structure calculations: DFT, MP2, etc.
QM/MM simulations
Large data visualization
- UMAP
- PMI-3D
- Properties plots
Properties calculation
- Tox/ADMET ML prediction (BBB, hERG, permeability (Caco-2, PAMPA etc.), metabolic stability prediction, etc.)
- PhysChem/MedChem properties prediction and analysis (logP/logD, solubility, etc.)
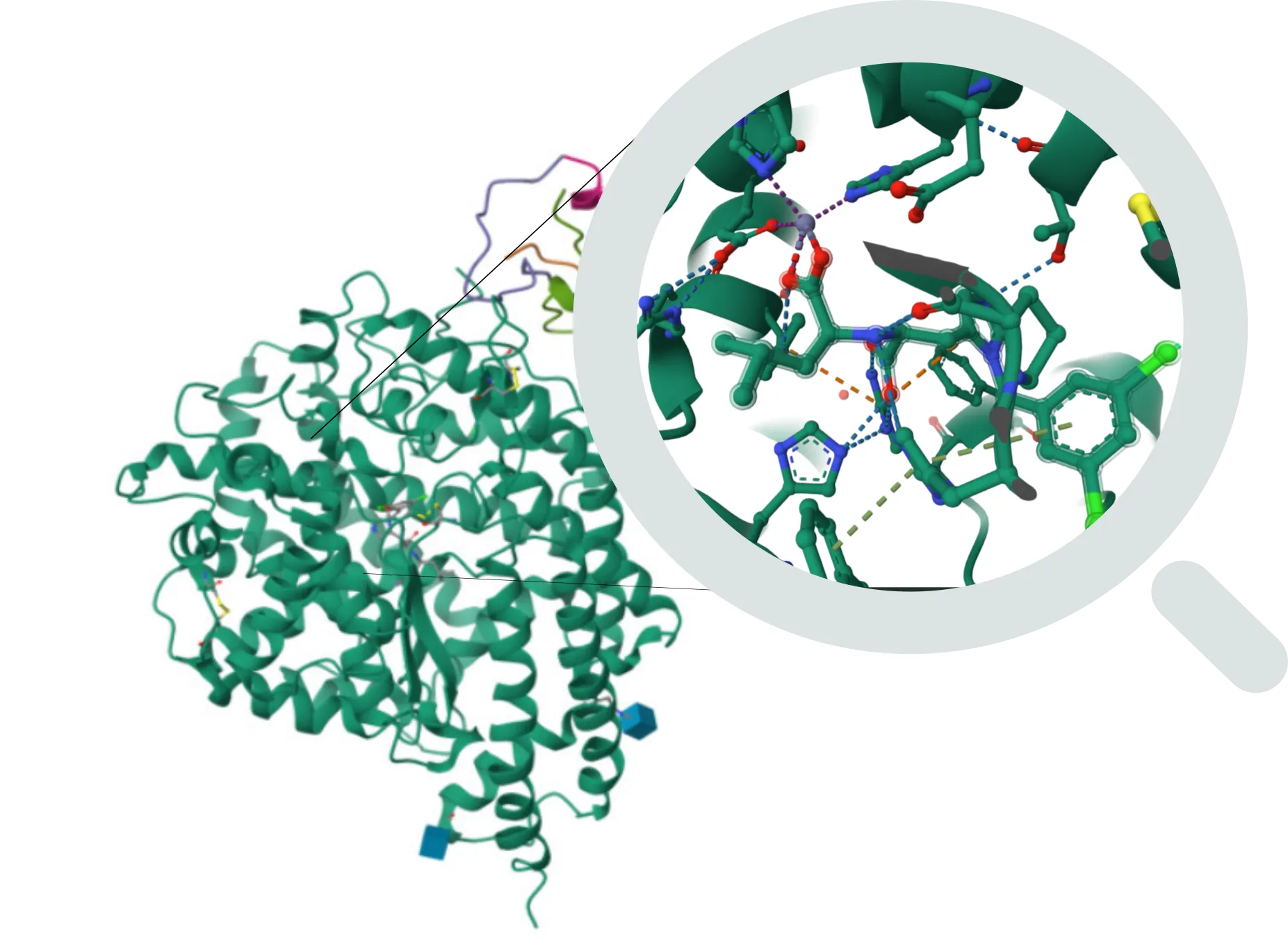
Structure-based virtual screening (SBDD) and Complexes Evaluation
Noncovalent molecular docking
- Decoys validated models
- Tailored constraints
- Multi-staged workflow
Covalent molecular docking
- Pre-programmed chemical reactions and designed targeted libraries
- Multi-staged workflow (+noncovalent docking for pre-reaction modeling)
Fragment-based virtual screening
Modeling assisted by Active Learning
Boltz-2 approach
- Structure prediction
- Determination of binding mode
- Estimated probability of binding
- Binding affinity prediction
Thompson Sampling
- Adaptive compound selection strategy
- Guided exploration of chemical space
- Efficient prioritization of diverse and promising candidates
Metadynamics
- Enhanced sampling technique for rare events
- Efficient identification of free energy minima and transition states
- Exploration of hidden conformational landscapes
- Supports binding pose refinement and mechanism elucidation
Molecular Dynamics (MD) simulation
- Validation of ligands selected during virtual screening
- Estimation protein-ligand binding energy
- Dynamic picture of protein-ligand interaction
QM/MM calculations
- Hybrid quantum–mechanical/molecular–mechanical modeling
- Accurate description of reaction mechanisms
- Refined modeling of covalent bond formation and charge transfer events
RAMD (Random Acceleration Molecular Dynamics):
- Predict ligand residence time (τ = 1/koff) from unbinding simulations.
- Reveal unbinding pathways and key interactions governing dissociation kinetics.
- Rank compounds by residence time to support kinetics-driven optimization and selectivity.
- Guide SAR and design decisions when affinity alone is no longer discriminative.
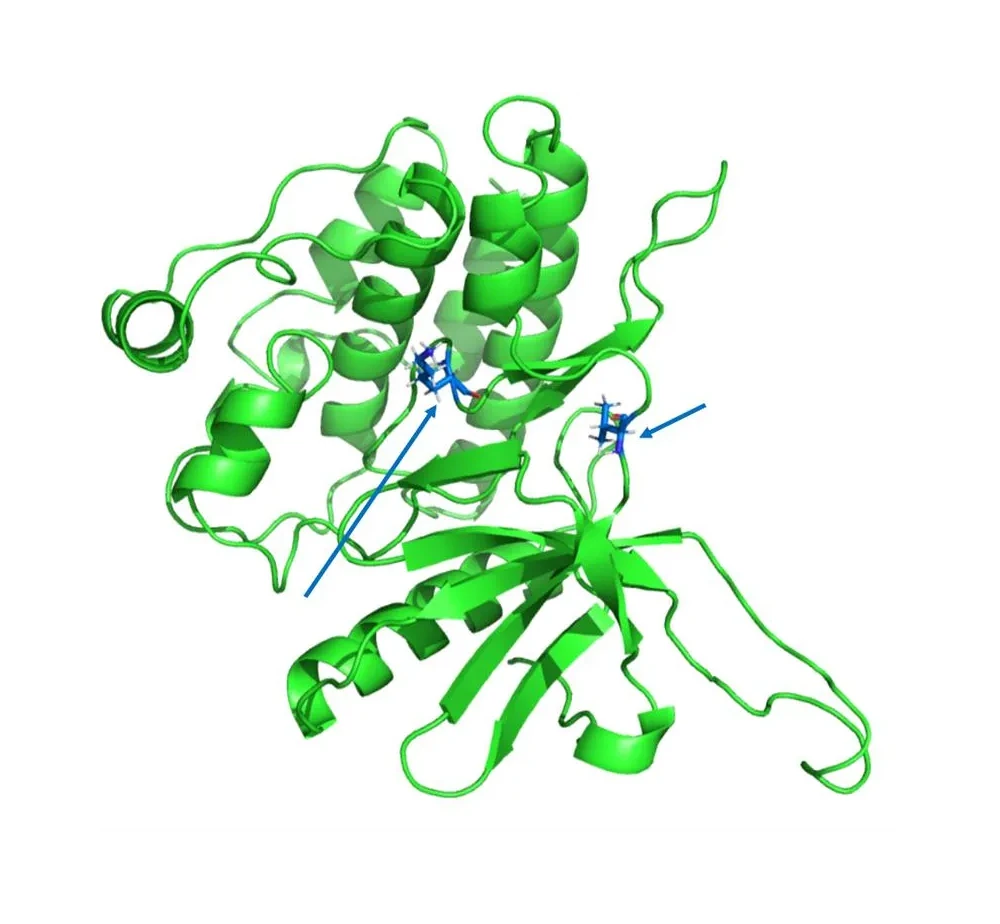
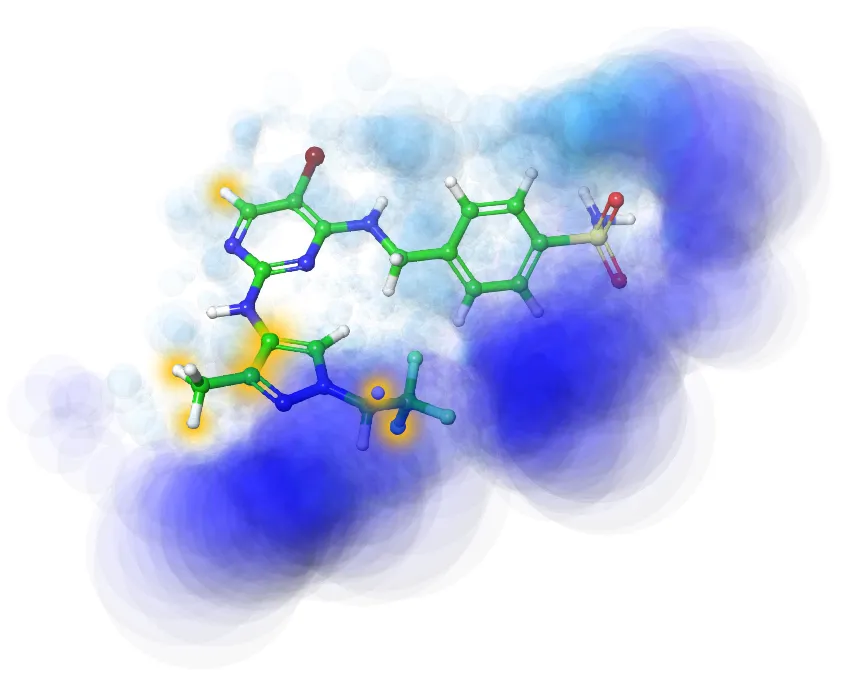
FEP (Free Energy Perturbation) - Precision Binding Affinity Prediction
What it solves:
- Accurate calculation of ligand binding affinity to the target
- Ranking compounds by binding free energy for improved SAR analysis
- Predicting the impact of modifications on binding affinity
- Reducing synthesis and biological testing costs by selecting the most promising candidates

Unlock a new level of accuracy in ligand design!
Every Project Starts with an Evaluation Phase
Before launching full-scale modeling, we always begin with a project evaluation phase — a concise analytical step designed to:
- define a scientifically legit strategy for virtual screening or lead optimization
- assess the availability and suitability of structural data (targets, ligands, etc.)
- recommend the most effective approach and scope tailored to your goals and constraints
- identify potential risks or challenges in early-stage drug discovery campaign
To explore a real example of our evaluation report to see the structure, level of detail, and types of insights you can expect:
Tailored to Your Project
We’d be happy to perform a custom evaluation specifically for your project — based on your target, available hits, or desired compound types. This is the best way to make confident decisions before committing to the next steps.
CADD-driven Biological Validation
- Minimal compounds delivery time (1-2 days)
- 25% off in case of access fee
- Custom workflow tailored to your needs
- 10% off for the CADD and biological services
- High chance to obtain up to 15 biologically validated hits for 35-75 working days
Project Portfolio Overview (2025)
Projects distribution across continents
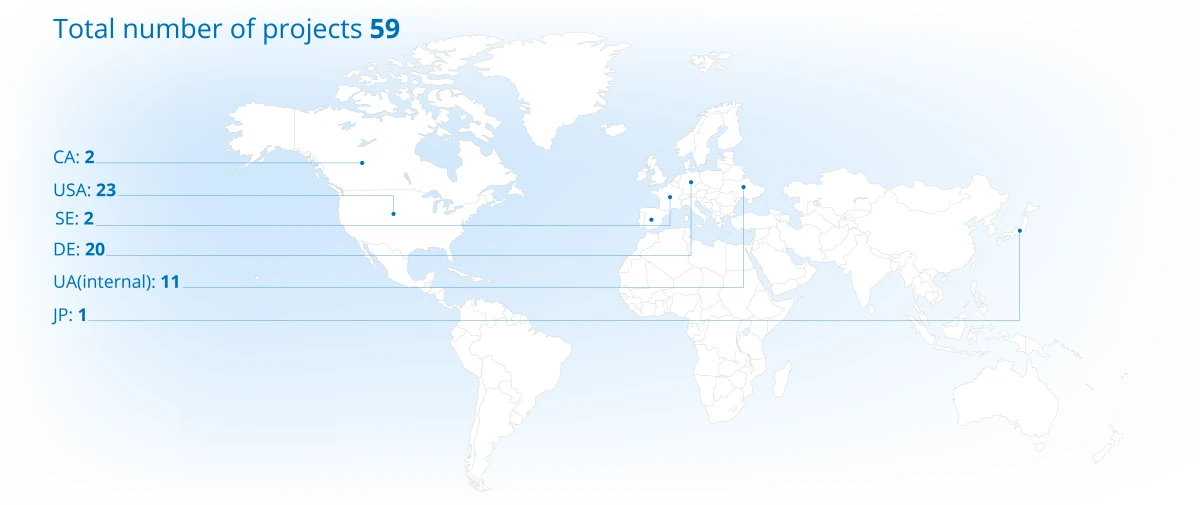
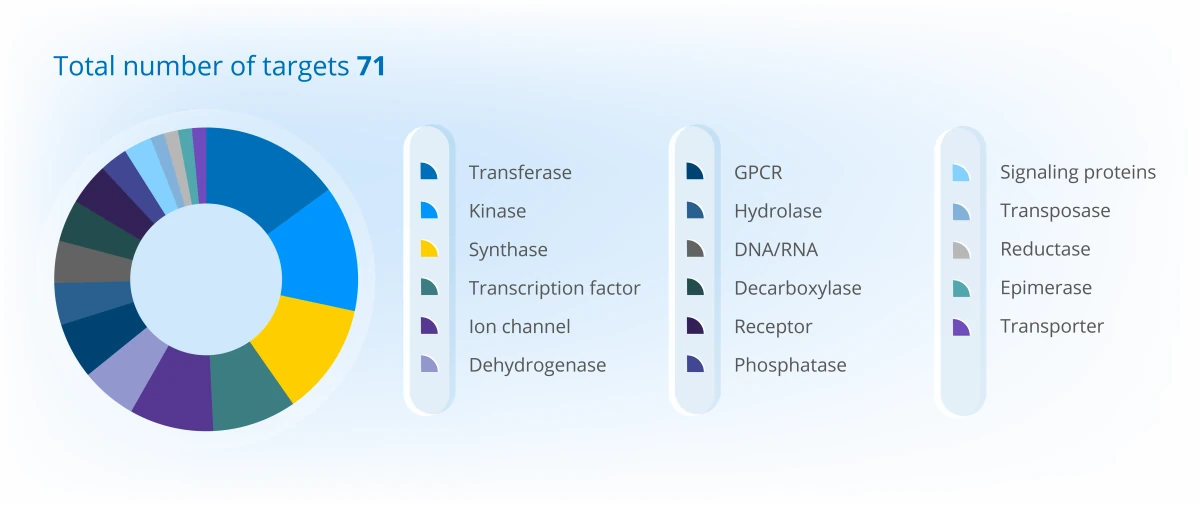

Discover the depth of our experience and success through specific case studies here.
Selected publications
- Neural Network Models for Prediction of Biological Activity using Molecular Dynamics Data: A Case of Photoswitchable Peptides
Molecular Informatics 2025, 44. DOI: 10.1002/minf.70001 - Structural flexibility and shape similarity contribute to exclusive functions of certain ATG8 isoforms in the autophagy process
Molecular Informatics 2025, 44. DOI: 10.1002/minf.70004 - Pharmacological inhibition of syntenin PDZ2 domain impairs breast cancer cell activities and exosome loading with syndecan and EpCAM cargo
Journal of Extracellular Vesicles 2020, 10. DOI: 10.1002/jev2.12039 - Structural Insight on the Selectivity of Calyx[4]Arene-Based Inhibitors of Mg2+−Dependent Atp-Hydrolases
Molecular Informatics 2025, 44. DOI: 10.1002/minf.202400200 - Alternative substrate-assisted hydrolysis pathways of posttransfer editing by prokaryotic leucyl-tRNA synthetase
The FEBS Journal 2025, 292. DOI: 10.1111/febs.70153 - Lysine Acetylation of Plant α-Tubulins: Scaling Up the Local Effect to Large System Transformations
Proteins: Structure, Function, and Bioinformatics 2025, 93. DOI: 10.1002/prot.26846 - Computational and experimental identification of putative αTAT1 modulators: implications for nervous system function
Frontiers in Pharmacology 2025, 16. DOI: 10.3389/fphar.2025.1654114 - Modelling of an autonomous Nav1.5 channel system as a part of in silico pharmacology study
Journal of Molecular Modeling 2021, 27. DOI: 10.1007/s00894-021-04799-w - Integrated workflow for the identification of new GABAAR positive allosteric modulators based on the in silico screening with further in vitro validation. Case study using Enamine's stock chemical space
Molecular Informatics 2024, 43. DOI: 10.1002/minf.202300156 - 4-(Azolyl)-Benzamidines as a Novel Chemotype for ASIC1a Inhibitors
Int. J. Mol. Sci. 2024, 25. DOI: 10.3390/ijms25073584


