We are thrilled to announce our collaboration between Enamine and ChemPass, which will open the way for significant advancements in drug discovery. ChemPass will apply its cutting-edge approach to design innovative fragment libraries, and Enamine ensures that these novel compounds are synthesized efficiently. Together, we are establishing a new standard for fragment-screening libraries that will enable scientists to do research more quickly and effectively.
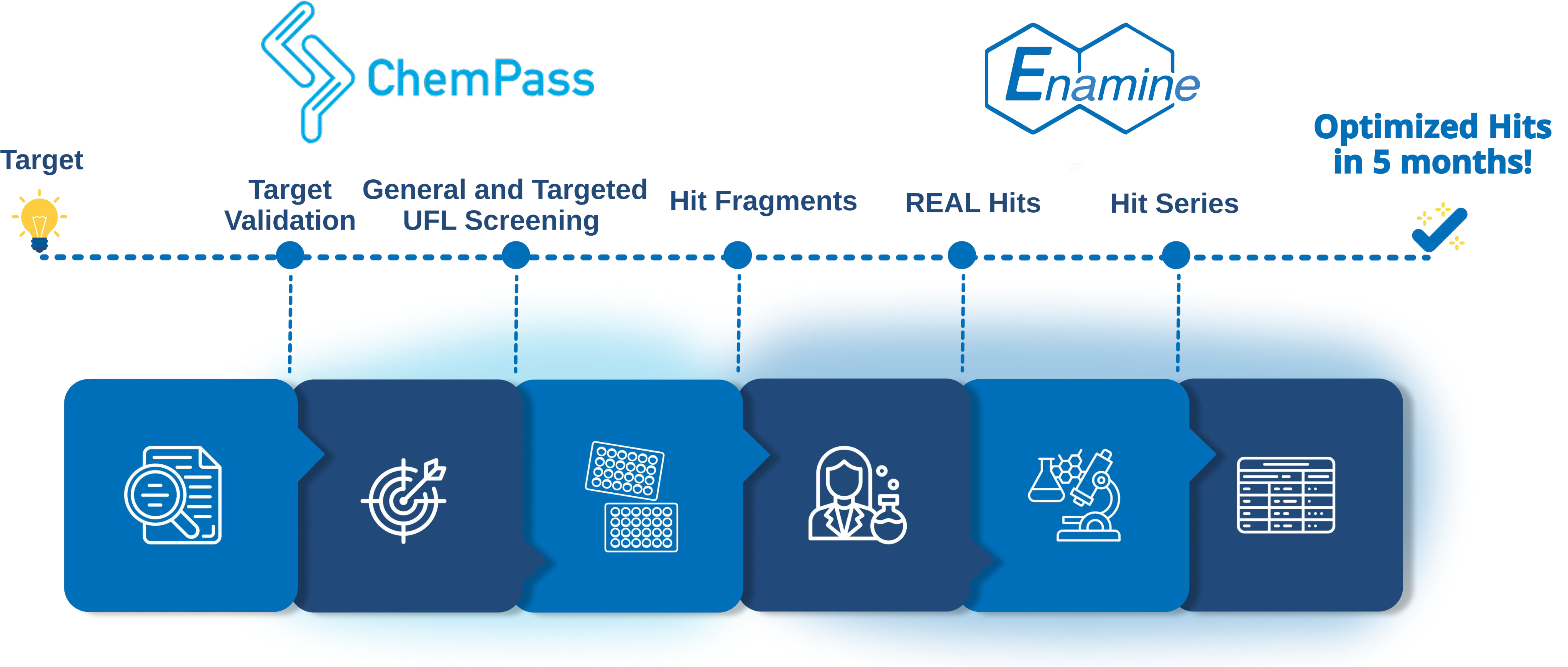
The joint effort between the two companies aims to significantly expand fragment-based hit discovery and to shorten the fragment-to-lead development cycle for challenging biological targets.
ChemPass, applying its Universal Fragment Library Design Platform, will provide general or target-specific fragment libraries based on the customer’s biological target of interest.
Enamine’s team will support the screening of the fragment libraries, the expansion of the obtained fragment hits into lead-like analogues from Enamine REAL® Space, the development of structure-activity relationship (SAR) by a few rapid rounds of screening of REAL® catalog compounds.
Overall, the application of the ChemPass Library Design Platform, Enamine’s REAL® combined with its extensive medicinal chemistry expertise, and Enamine’s screening facilities in discovery biology will deliver optimized hits to our clients in five months, a true acceleration of early-phase drug discovery.
Why UFrag™ library from Enamine & ChemPass?
- Higher success against tough targets.
- Less redundancy for easy targets.
- Superior pharmacophore analysis – better coverage, higher hit rates, fewer redundant compounds.
- Optimized fragment growth – improved hit rates and optimizability.
- More efficient vector analysis – fewer compounds, faster drug discovery.
- Short synthesis timelines, low costs, and high laboratory success rates.

Enhance the performance of your fragment library with our Service.
Leveraging our advanced fragment design platform, ChemPass and Enamine will conduct a comprehensive analysis of your existing library to identify gaps and recommend strategically selected fragments or stock solutions. This approach ensures broader binding site coverage, improved hit rates, and greater efficiency in screening campaigns, including those targeting challenging proteins. Alternatively, a general and comprehensive UFrag™ library is available from Enamine.
Optimize your fragment library for more effective and successful drug discovery!

Why UFrag™ library from Enamine & ChemPass?
- Higher success against tough targets.
- Less redundancy for easy targets.
- Superior pharmacophore analysis – better coverage, higher hit rates, fewer redundant compounds.
- Optimized fragment growth – improved hit rates and optimizability.
- More efficient vector analysis – fewer compounds, faster drug discovery.
- Short synthesis timelines, low costs, and high laboratory success rates.
The effectiveness of the hit identification process depends on the screening method and screening library quality. Typically, diversity analysis is utilized to select currently available covalent and noncovalent libraries with desirable properties. To select current covalent and noncovalent fragment libraries with desirable properties, diversity analysis is typically utilised. ChemPass' UFrag platform is based upon pharmacophore, shape and vector analysis of vast ligand binding data in the protein database and it combines several key technologies that are necessary to revolutionise fragment library design.
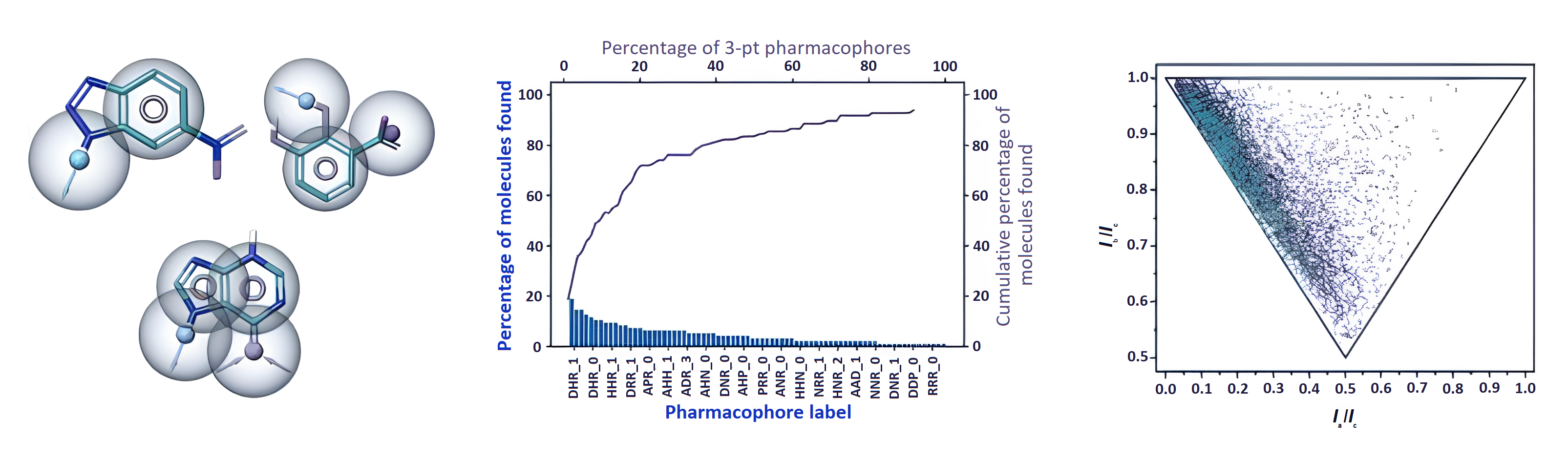
Conventional fragment libraries created through diversity analyses have been demonstrated to contain a significant number of redundant spacer and pharmacophore motifs, which leads to few to no hits against pockets of challenging targets and high hit rates of redundant rings for specific targets. The outcome is a significant danger of missed chances and a very varied experimental hit rate.
The platform's inherent customisation features allow the library design to target particular protein classes while minimising both the size of the library and the expense of synthetic materials. In order to confirm the notion, a test library was screened against a validated and hard target. The results showed that the test library produced a much higher hit rate and binding site coverage against the hard target than a standard fragment library.
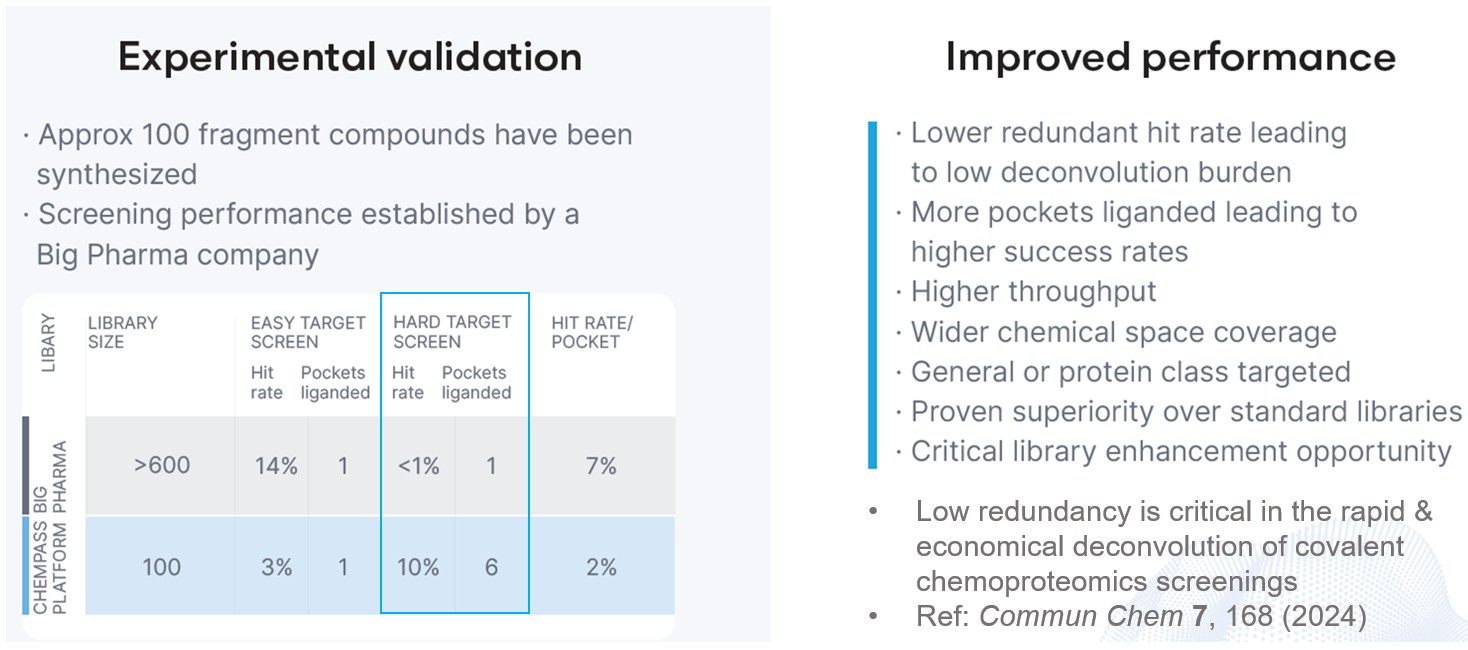
Why UFrag™ library from Enamine & ChemPass?
- Higher success against tough targets.
- Less redundancy for easy targets.
- Superior pharmacophore analysis – better coverage, higher hit rates, fewer redundant compounds.
- Optimized fragment growth – improved hit rates and optimizability.
- More efficient vector analysis – fewer compounds, faster drug discovery.
- Short synthesis timelines, low costs, and high laboratory success rates.
A new fragment design platform!
We are thrilled to announce our collaboration between Enamine and ChemPass to advance fragment-based drug discovery. ChemPass will apply its Universal Fragment Library (UFL) Design Platform to create target-specific fragment libraries, while Enamine will contribute with efficient synthesis and screening. This collaborative initiative will accelerate hit finding and the hit-to-lead process for our customers that seek to shorten their early discovery cycles.
UFL
A Universal Fragment Library designed using vast protein-ligand binding knowledge to enhance hit discovery.
Enhancement with UFrag™
Optimize your fragment libraries for better hit rates, binding coverage, and efficient screening.
Fragment Screening Service
A streamlined screening process combining fragment screening, computational tools, and vast chemical space.

Tailored towards your needs!
Discover new horizons with Enamine Computer-Aided Drug Discovery services. Partner with our skilled team, experts in crafting specialized workflows using cutting-edge chemoinformatics and modeling tools. We believe that teamwork among chemists, biologists, and chemoinformaticians is key to success. That's why our departments work closely together to improve the efficiency of your drug discovery project significantly. Our CADD team offers a comprehensive solutions to navigate you through Enamine Collections, ensuring efficient hit finding and hit-to-lead optimization.
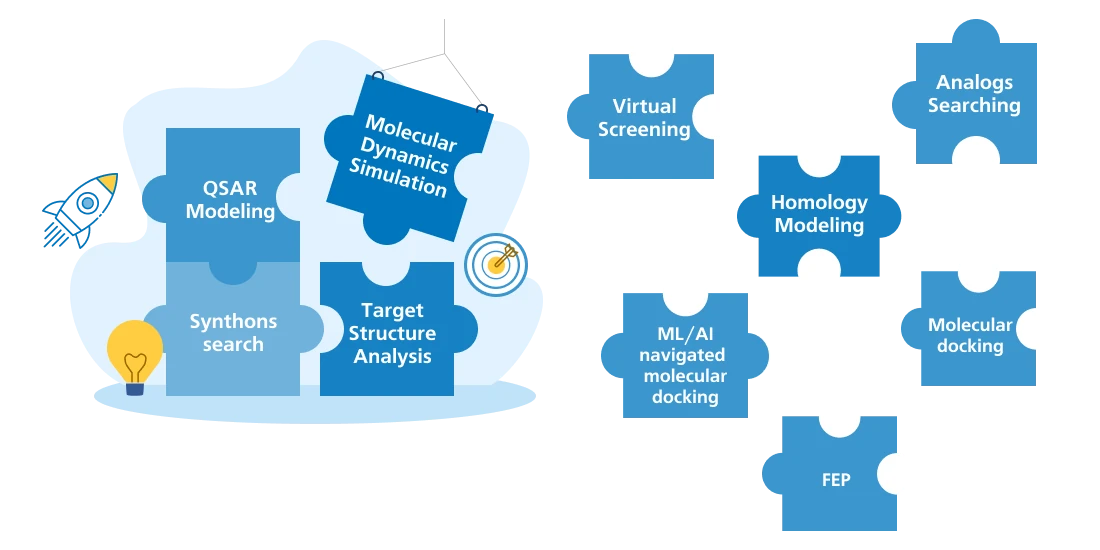
Target Modeling
Molecular Dynamics (MD) Simulation
- Comparison and demonstration of the behavior of structures over time with and without a ligand
- Modeling the impact of mutations on stability, activity, and resistance to known binders
- Validation of ligands selected during virtual screening
- Estimation protein-ligand binding energy
- Cryptic pocket searching
Homology Modeling
- 3D-structure prediction for unresolved targets
- Structure repairing
- Mutant modelling
Target Structure Analysis
- Potential binding site searching
- Active residue estimations
DNA/RNA - Complex Modeling
- Intercalation and binding in RNA groves
- G-quadruplex modeling
- Ryboswitches
- DNA GG and GA binding pocket
Ligand Searching
Analogs Searching
- 2D searching (Morgan Fingerprints, ECFPs, etc.)
- 3D searching (Electroshape, USRCAT, E3FP, etc.)
- Diversification
- Search of BM scaffolds, substructure search, synthons search
Virtual Screening
- Noncovalent molecular docking
- Covalent molecular docking
- 3D pharmacophore screening
ML Modeling and Prediction
- ML-powered high-throughput virtual screening
- ML-facillitated building of QSAR models
- ML-driven ADMET properties prediction
Hit-to-Lead Integrated Projects Support
- Evaluation of synthetic feasibility and screening result analysis
- Re-scaffolding
- Bioisosteric replacement
NEW APPROACH
FEP (Free Energy Perturbation) - Precision Binding Affinity Prediction
What it solves:
- Accurate calculation of ligand binding affinity to the target
- Ranking compounds by binding free energy for improved SAR analysis
- Predicting the impact of modifications on binding affinity
- Reducing synthesis and biological testing costs by selecting the most promising candidates

Unlock a new level of accuracy in ligand design!
We use a balanced approach employing different methods complementing each other
CADD-driven Biological Validation
- Minimal compounds delivery time (1-2 days)
- 25% off in case of access fee
- Custom workflow tailored to your needs
- 10% off for the CADD and biological services
- High chance to obtain up to 15 biologically validated hits for 35-75 working days
Hit-to-Lead Optimization
- Comparison and analysis of physicochemical properties of actives vs. inactives
- Clustering of active and inactive compounds for further analysis
- Building a docking model and screening pipeline using the experimental biological data and validation with active and inactive structures
- Actives vs. inactives: a pairwise comparison between structurally similar molecules to identify key elements that can be used as discrimination factors
- MD simulation of complexes to identify interaction changes over time
Project Portfolio Overview (2024)
Projects distribution across continents
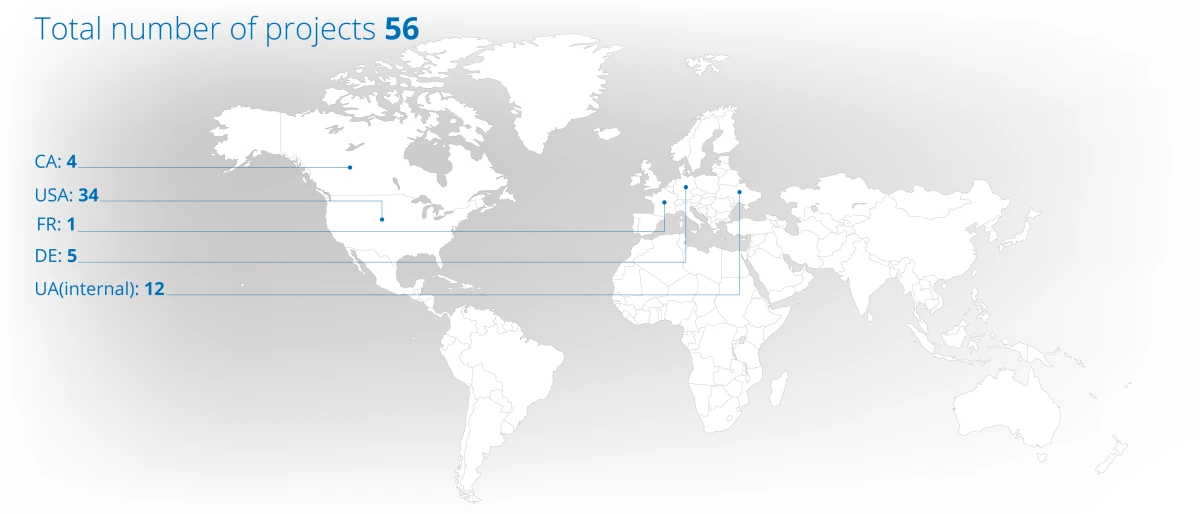


Discover the depth of our experience and success through specific case studies here.
Selected publications
- Creation of targeted compound libraries based on 3D shape recognition.
Mol Divers 2022, 27, 939–949. DOI: 10.1007/s11030-022-10447-z - Pharmacological inhibition of syntenin PDZ2 domain impairs breast cancer cell activities and exosome loading with syndecan and EpCAM cargo.
J of Extracellular Vesicle 2020, 10. DOI: 10.1002/jev2.12039 - Modelling of an autonomous Nav1.5 channel system as a part of in silico pharmacology study.
J Mol Model 2021, 27. DOI: 10.1007/s00894-021-04799-w - Integrated workflow for the identification of new GABA positive allosteric modulators based on the in silico screening with further in vitro validation.
Molecular Informatics 2023. DOI: 10.1002/minf.202300156 Case study using Enamine’s stock chemical space.