Designed for discovery of new water channels modifiers
1 348 compounds
The library was designed to find molecules which target both end-chains of water channels (Aquaporins) AQ1, AQ4, AQ5 monomers and internal central pore of AQ5, that possess same physico-chemical properties as those in AQ1 and AQ4. A small number of known active inhibitors against this type of targets do not allowed us to build qualitative QSAR model or pharmacophore models. The HTS docking approach was used to search of new potential inhibitors, 2D similarity search was carried out to enrich the library with compounds similar to reported actives.
Download SD files
1 348 compounds for cherry-picking
Library Design
Docking optimization & calculations
Three protein structures (3D9S, 1IH5, 3GD8) were chosen after analysis of 11 available X-Ray structures (1H6I, 1IH5, 2ZZ9, 3GD8, 4NEF, 3D9S, 5DYE, etc.). However, it is known that shift in open/closed state is dramatically sensitive to some mutations. This data described several hot-spots, which are located inside the channel and were used for preparation of docking models on the primary stage of protein structure refinement, of course if these residues are involved in interaction, not in rigidity of the helix. Additionally the whole surface of each monomer was probed with FT-MAP server, which showed itself as a very accurate and trustworthy tool for binding site identification. It is based on evaluation of energy interaction between small organic probes (solvents, saturated and unsaturated cycles...) and the surface of interest.
The design of aquaporin library was performed with implementation of two different approaches: flexible docking and high throughput screening. Flexible docking protocol assumed a partially-free rotatable motion of side-chains, which are located in the entrance of both cavities in AQ5/AQ4/AQ1 proteins. The area of flexibility covered about 5 Å2 around the centre of the channel in one unit of the tetrameric structure. The centre of a Grid box (determination of the binding area) was selected based on residue mutation studies and FT-MAP calculations, mentioned above. To expand the volume of putative binding sites a set of real inhibitors, which were disclosed in publications and chemblDB, were docked together with compounds, which were shown to interact with the target (AqB013, Bumetanide, Zonisamide, Arbidol and Acetazolamide were taken from the literature and some neurotransmitters, like dopamine, epinephrine and serotonine, were chosen based on our collaboration with academicians.
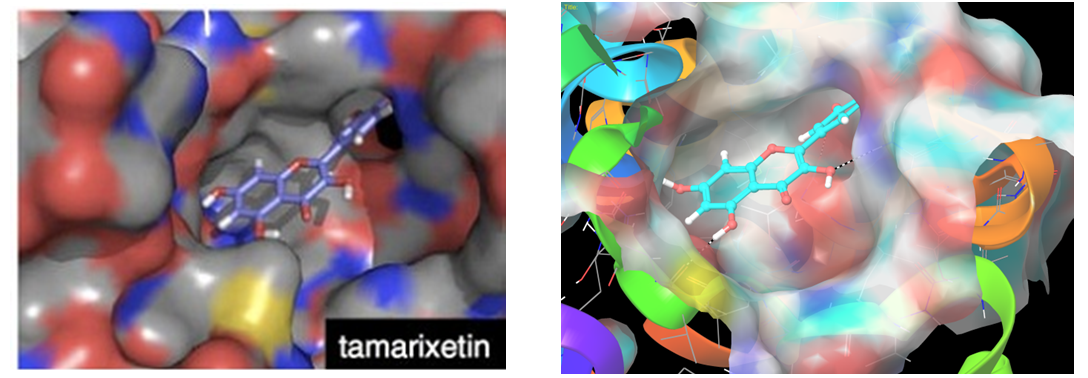
Reported binding mode of tamarixetin (left) and representation of hit molecule obtained after our docking calculations (right).
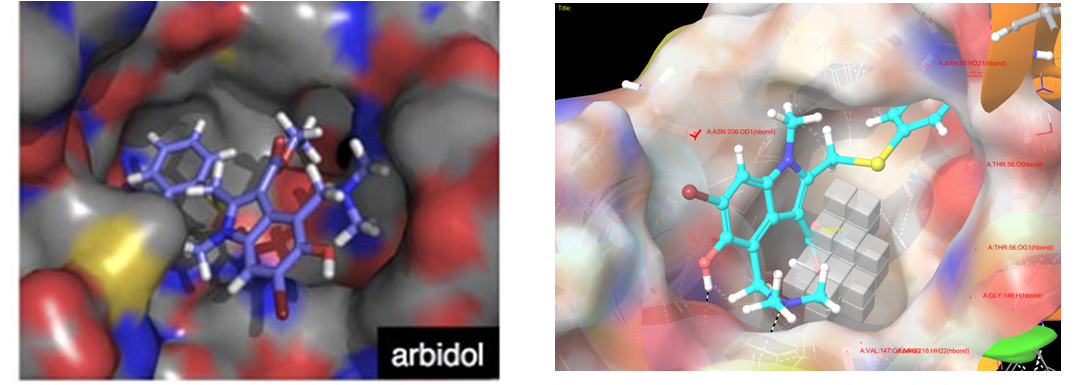
Several residues, which are responsible for substrate/inhibitor binding - G146, V147, R216, T56, (Asn58, Gly209, Val141), are also important for arbidol binding.
In this way a more suitable conformation of the gate was achieved in binding sites and these optimized structures were applied for screening procedure with slightly less exhaustive parameters of pose estimation and number of generated conformers. Here it is a short example of parametrization if the docking into the AQ5 model. Among constraints, which formed the filtering sieve, a hydrophobic core inside the channel and partial match against the set of amino acids (ARG:188, ASN:120, GLY:181, A:HIS:173, VAL:119 for AQ5 site2; and GLN:81, HIE:67, PRO:62, SER:152, THR:150, THR:150, TYR:90, TYR:90 for AQ5 site1), should be mentioned. All of them are shown in the figure. Those compounds which possessed a hydrophobic moiety and satisfied another condition (2-3 h-bond interactions with any of the listed residues), reached the next stage of more accurate and time-consuming docking procedure. Rating of these compounds is a combination of empiric values, post-processing calculation of energy, steric and h-bonding impacts. The more higher rating, the more probable binding of the ligand.
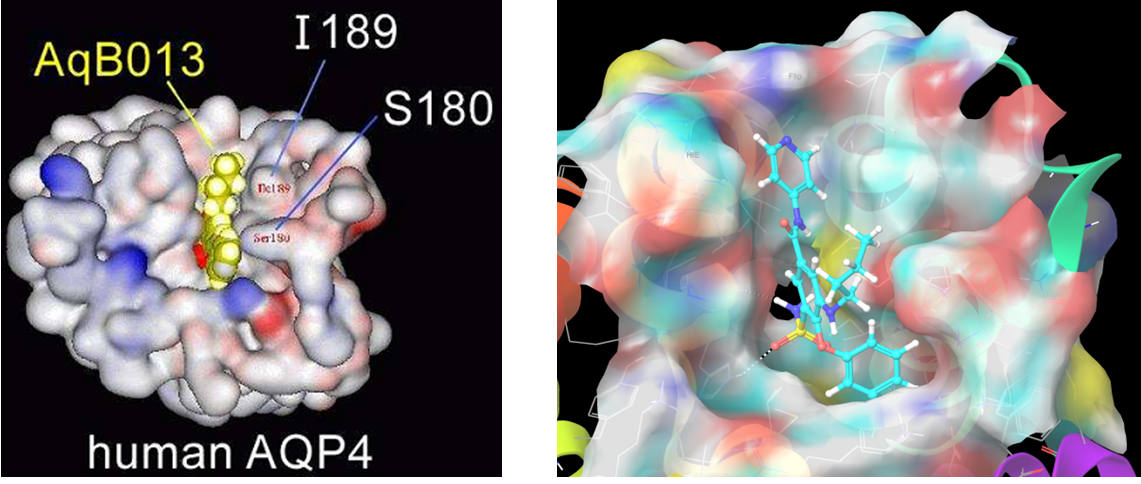
Compound AQB013 (the same for Bumetanide) docked in the second site of AQ4. The more preferable residues set is I189, S180, Phe175, Arg261, which are forming the opposite gate of the AQ4 channel.


