Designed for discovery of new Nav1.7 channel blockers
5 440 compounds
Sodium voltage-gated ion channel considered to be an important component in nociception. Therefore, selective Nav1.7 channel blockers are considered as important novel analgesics.
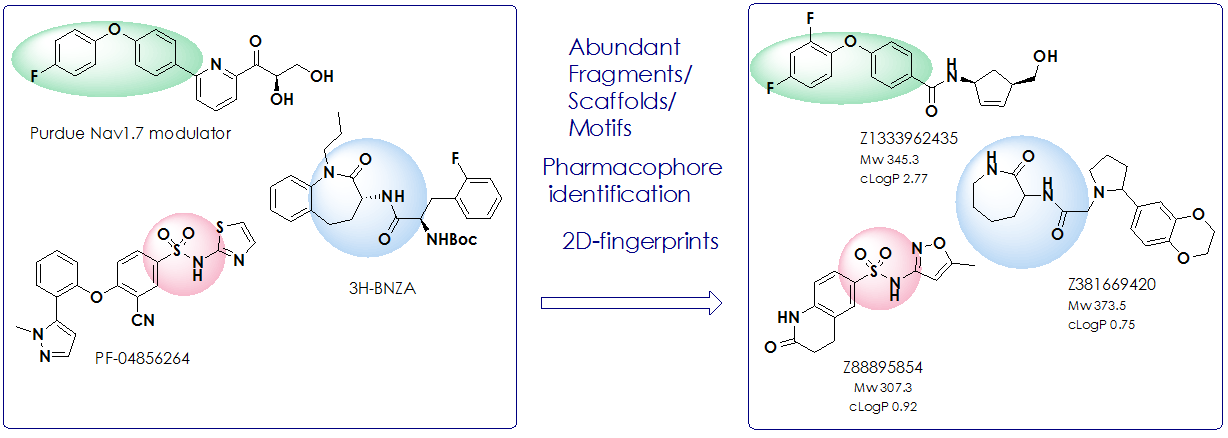
Analyzing the structures of recently developed ligands several main features have been identified:
- the most abundant are relatively flat aromatic cores;
- presence of highly polar backbone fragments/moieties (e.g. CONH2, SO2NHR, polar heterocycles, etc);
- compounds lay squarely in the middle of drug-like chemical space.
A set of substructure queries and 2D-fingerprintes based on found common structural motives and pharmacophores was used in searching Enamine screening collection to result in 5 440 compounds after application of medchem filters. The Nav1.7 Targeted Library is rich in compounds bearing saturated backbones that can be often seen in structures of other ion-channel blockers. All compounds meet requirements of Ro5, and 67% compounds are considered lead-like.
Download SD file
Library code: SICL-5440
Version: 24 May 2020
5 440 compounds
sublibrary of ICL-36
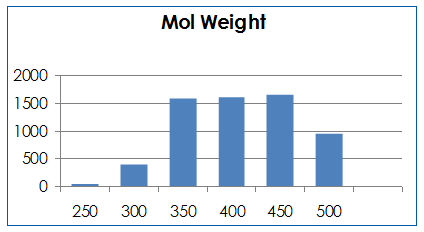
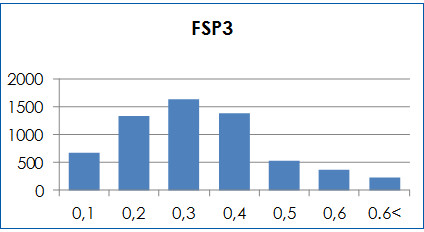
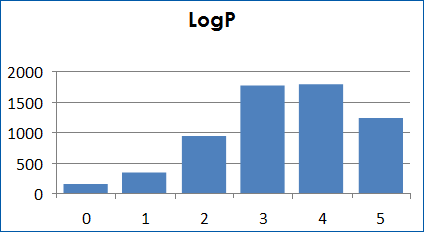
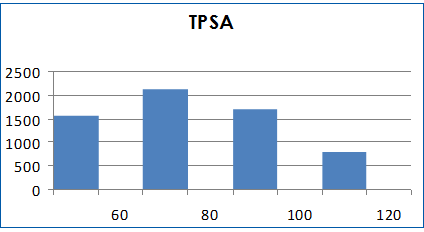
Molecular properties
Designed for discovery of new Voltage-gated calcium channel blockers
10 560 compounds
Calcium ion channels are responsible for an unusually large variety of physiological functions. Calcium ions entering the cell through voltage-gated channels serve as the second messenger of electrical signaling, initiating many different cellular events. As a target class calcium channels offer both challenges first of in designing selective antagonists of the channel subtypes and great opportunities following proof-of-concept provided by the marketed drugs.
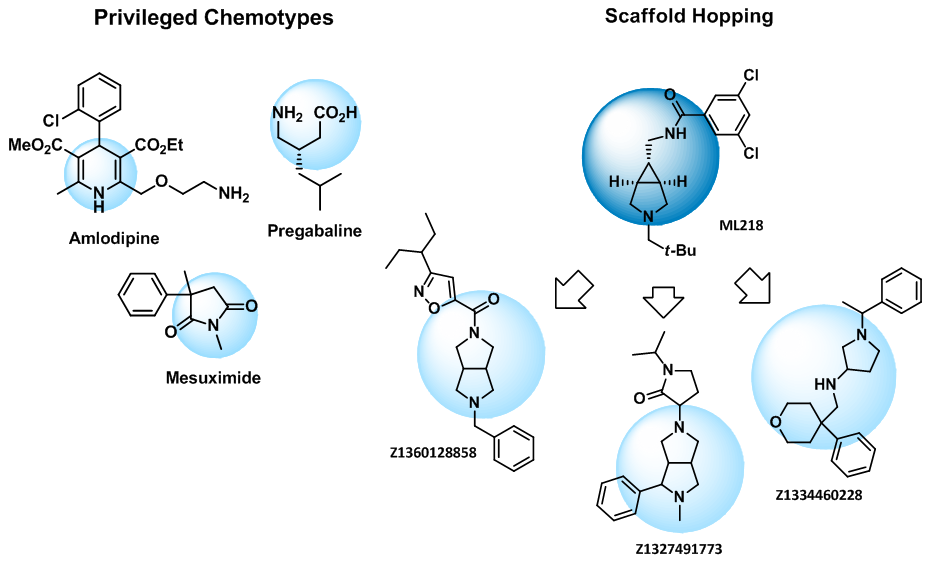
Enamine voltage-gated targeted library encompasses both known chemotypes and molecules based on novel scaffolds identified in our in silico studies and MedChem based scaffold hopping projects.
Download SD files
Library code: CICL-10560
Version: 24 May 2020
10 560 compounds
sublibrary of ICL-36
Main futures of the library:
- Lead-likeness: 86%
- Average Fsp3 0.38; average TPSA 60.5 Å2
- Novel Chemotypes
Library design
We have undertaken in-depth analysis of known calcium channel blockers and carefully selected 10 560 most promising drug-like structures from our screening collection exceeding 4.7M+ small molecular weight compounds.
Privileged chemotypes such as dihydropyridines, GABA-analogues, succinimides, 4-substituted prolines, and quinazolines were included in the library.
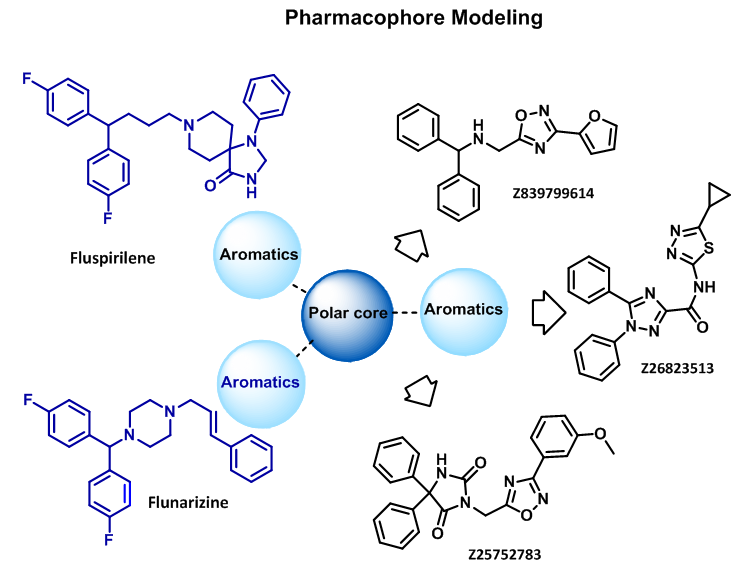
Pharmacophore modeling was carried out for several relevant calcium channel blockers resulting in about 1 700 compound set.
Scaffold hopping Structures of numerous known calcium channel blockers contain piperazine or 4-aminopiperidine. Recently several successful examples of bioisosteric replacement of these motifs by other diamine cores have been reported. We have extended this approach to a rich variety of compounds built on unique analogues of piperazine and 4-aminopiperidine available exclusively from Enamine and continued with other cores, e.g. imidazopiredazines, providing novel sets of compounds.
Molecular properties Drug like compounds with the most attractive structural and physical chemical properties. HBond Donors 0… 4, HBond Acceptors: 1 … 8, Rotatable Bonds: 0 ... 10, TPSA 3 … 140 Å2
Since 2016 Enamine synthesize over 15 000 new covalent compounds each year. This greatly contributes to the constant updating of our libraries in line with the latest trends in covalent warheads.
Product catalog
CSL-11760
Size
11 760
compounds
Description
Diverse covalent library with most demanded warhead types
Download file
Covalent Serine Hydrolase Library
CSHL-12160
Size
12 160
compounds
Description
Designed for discovery of mild electrophilic inhibitors of the largest enzyme class
Download file
Coronavirus Mpro covalent Library
MPC-2640
Size
2 640
compounds
Description
Designed for the discovery of new SARS-CoV-2 and pan-Coronavirus antivirals
Download file
CFL-8480
Size
8 480
compounds
Description
Diverse covalent warheads with balanced reactivity
Download file
Cysteine-Focused Covalent Library
CYS-3200
Size
3 200
compounds
Description
Library of Cys-specific covalent electrophilic binders
Download file
Serine-Focused Covalent Library
SER-1600
Size
1 600
compounds
Description
Special selection of Serine focused irreversible binders
Download file
Lysine-Focused Covalent Library
LYS-1600
Size
1 600
compounds
Description
The ultimate selection of Lys-specific binders
Download file
Electrophilic Covalent Probe Library
ECPL-960
Size
960
compounds
Description
Characterized by a new HTS thiol-reactivity assay
Download file
sACR-4160
Size
4 160
compounds
Descriptions
Diverse screening Acrylamides pre-plated at 10 mM concentration
Download file
fACR-2240
Size
2 240
compounds
Descriptions
Representative selection of fragment Acrylamides pre-plated at 100 mM stock concentration
Download file
sCLA-1200
Size
1 200
compounds
Descriptions
Library of diverse HTS-size chloroacetamides pre-plated at 10 mM concentration
Download file
Chloroacetamide Fragment Library
fCLA-1360
Size
1 360
compounds
Descriptions
Diverse strict Ro3 compliant chloroacetamides plated at 100 mM stock concentartion
Download file
SFF-640
Size
1 120
compounds
Descriptions
Representative selection of N-, O-linked and Aryl sulfonyl fluorides within fragment space
Download file
Size
960
compounds
Description
Enantiomeric pairs of covalent electrophilic fragments
Download file
CMF‑141
Size
141
compounds
Description
Covalent Heterocyclic Fragment Library for identification of Cryptic and Allosteric Pocket
Download file
Size
960
compounds
Description
2-Sulfonylpyrimidines for Next-Generation Covalent Targeting
Download file
Support
- Hit Confirmation: QC check, HPLC repurification, resynthesis
- Hit follow-up: analogs search from stock or REAL Database
- Fast hit exploration libraries synthesis
We offer comprehensive support in developing your hit compounds. Naturally such programs are realised most efficiently when biological actives originate from our screening collection. However, even if the hit compounds are from the collections of other vendors lead identification and optimization projects can proceed most productively in our hands. Sometimes for this we only need to synthesize first examples of the given chemical series and validate synthesis route.
Enamine Fragment Collection currently contains 259 380 fragments being the largest and most reliable source of quality fragments. A number of focused fragment libraries were designed to perfectly meet needs of our clients. We collaborate with the leading experts in FBDD field on design and supply of top fragment libraries.
ESS-320
Size
320
compounds
Description
Elaborated tool for initial screen
Download file
High Fidelity Fragment Library
HFF-1920
Size
1 920
compounds
Description
Fragments of high MedChem tractability
Download file
DSI-860
Size
860
compounds
Description
Designed for easy and rapid follow-up synthesis
Download file
MiniFrags-80
Size
80
compounds
Description
Guiding optimisation of fragment-derived lead compounds
Download file
CFL-8480
Size
8 480
compounds
Description
Diverse covalent warheads with balanced reactivity
Download file
FFL-d6
Size
1 280
compounds
Description
Specially designed for 19F NMR ligand-based screening
Download file
Size
800
compounds
Description
Designed for easy and efficient exploration of novel protein targets
Download file
Natural Product-like Fragments
NPL-4160
Size
4 160
compounds
Description
Source of biologically validated starting points
Download file
3D Shape Diverse Fragment Library
3DF-1200
Size
1 200
compounds
Description
Unique 3D diversity among shaped molecules
Download file
PPIF-3600
Size
3 600
compounds
Description
Fragments able to mimic protein structural motifs and hot-spot residues
Download file
Single Pharmacophore Fragments
SPF-1500
Size
1 500
compounds
Description
Fragments for easy-to-analyse protein-ligand interaction
Download file
Carboxylic Acid Fragment Library
CAF-4000
Size
4 000
compounds
Description
Designed for specific protein targets and sensible onset
Download file
Size
1 280
compounds
Description
The most medchem reliable source of carboxylic acids replacement
Download file
Halogen-enriched Fragment Library
HEF-1920
Size
1 920
compounds
Description
Library of high diversity of halogen bonding motifs
Download file
Electrophilic Covalent Probe Library
ECPL-960
Size
960
compounds
Description
Characterized by a new HTS thiol-reactivity assay
Download file
Covalent Heterocyclic Fragment Library
CovHetLib‑141
Size
141
compounds
Description
Covalent Heterocyclic Fragment Library for identification of Cryptic and Allosteric Pocket
Download file
CNSF-1
Size
1 280
compounds
Description
CNS-friendly molecules capable of BBB penetration
Download file
Size
960
compounds
Description
Enantiomeric pairs of covalent electrophilic fragments
Download file
Size
372
compounds
Description
Size-optimized fragment library efficiently covering pharmacophore space, developed using ChemPass's Universal Fragment Library (UFL) Design Platform.
Download file
Size
2 301
compounds
Description
General fragment library developed with ChemPass's Universal Fragment Library (UFL) Design Platform.
Download file
Support
- Hit Confirmation: QC check, HPLC repurification, resynthesis
- Hit follow-up: analogs search from stock or REAL Database
- Fast hit exploration libraries synthesis
- Fragment growing and linking within Enamine REAL and Chemspace Freedom spaces
We offer comprehensive support in developing your hit compounds. Naturally such programs are realised most efficiently when biological actives originate from our screening collection. However, even if the hit compounds are from the collections of other vendors lead identification and optimization projects can proceed most productively in our hands. Sometimes for this we only need to synthesize first examples of the given chemical series and validate synthesis route.
Modern drug discovery requires contemporary compound libraries. We synthesize 300,000 new compounds annually and select the best candidates for our compound libraries. We offer over 100 fast-to-deliver pre-plated compound libraries for various research needs, from large-scale HTS screens to assay-specific and proteomics studies.
- Positive screening outcome. We conduct in-depth analysis of the research field and collaborate with leading industry experts to design our libraries.
- Premium quality. We produce compound libraries using only over 90%-pure compounds with analytics (LCMS and/or NMR) not older than one year. We filter compounds that are unstable in DMSO, have poor solubility, and undesirable salts. We also track the number of freeze-thaw cycles to determine pre-plated libraries shelf life based on their nature (cf. covalent vs non-covalent and other sensitive classes). When resupplying the hits, we follow our defined procedure, tracking the compound batches and their analytics. We purify or even resynthesize samples free of charge if they are of poor quality.
- Resupply from dry powders. For the library preparation, we use compounds available at 10+ mg in our stock so that they can be resupplied from the same batch. We have a program in place to maintain the dry sample stock levels through resynthesis for compounds present in REAL Space. For the best and fastest results, we encourage integrated lead discovery projects involving our biological screening, computational chemistry, efficient Hit-to-Lead using our REAL Space, ADMET/PK, and MedChem support.