(This post originally appeared on LinkedIn)
Notoriously toxic, covalent inhibitors have been nearly excluded from major drug discovery programs in the not so distant past. Ironically, a great amount of the important drugs exploit covalent inhibition as their mechanism of action (MOA). The view on covalent inhibitors is shifting towards a wider consideration, however, inspired by recent successes with EGFR inhibitors for treating cancer and many other promising examples. A recent publication in Nature provides a chemical proteomic platform for the global and quantitative analysis of lysine residues in native biological systems, offering further insights and ideas in this area.
Historically, covalent inhibitors have been discovered either through serendipity, for example omeprazole, or as derivatives of natural products, such as amoxicillin -- the β-lactam antibiotic derived from penicillin.
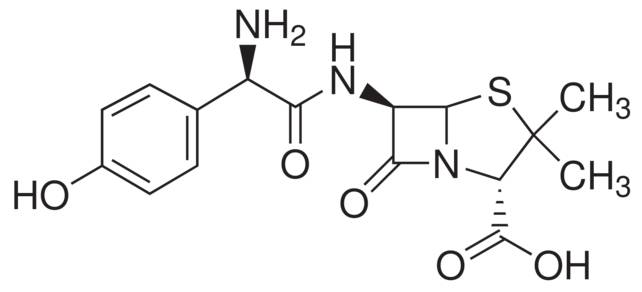
A structure of Amoxicillin
However, the current renaissance of covalent inhibitors has been fueled by the continuing advancement of medicinal chemistry approaches, and rational drug design. Equipped with increasingly available crystal structure information about different small molecule-protein complexes, researchers are able to modify high affinity reversible ligands with sparingly reactive electrophiles and building more efficient molecules.
Filled with hopes and disappointments, the viability of covalent inhibitors as an efficient strategy to fight certain life threatening conditions, like Non-Small Cell Lung Cancer, is still to be proved in future research programs. What is clear now, though, is that targeted covalent inhibitors appear to be a highly underappreciated opportunity in modern drug discovery research, attracting a growing interest among medicinal chemists in academia and pharma.
Pursuing alternatives: a focus on Lysine
While the overwhelming majority of currently reported covalent inhibitors targets a nucleophilic cysteine residue in close proximity to the ligand-binding site, this strategy is limited due to the low abundance of the latter in the overall proteome. In a pursuit of viable alternatives, a number of other nucleophilic residues have been targeted, including serine, tyrosine, threonine, aspartate, and lysine. A recent review in Angewandte Chemie International Edition is focused specifically on lysine as a potentially viable approach to covalent inhibition. Taking into account the basicity of the ε-amino group in lysine, drug discovery research around this target is associated with a number of specific challenges. The authors summarize the key principles for lysine-targeting inhibitor design, give historical examples and present recent developments that demonstrate opportunities open for future drug discovery efforts in this direction.
Quality vs quantity
The rational design of lysine targeted covalent inhibitors is a challenging task, largely facilitated by the availability of carefully selected compound libraries. On the other hand, a fragment based approach seems to be a powerful strategy to develop covalent inhibitors targeting lysine residue. With that in mind, Enamine chemists created a Lysine-focused covalent fragments library, containing 1,600 quality fragments, which can be a valuable tool for probing grounds in this area.
The common electrophiles, such as vinyl sulfones and sulfonamides, acrylamides, β-(dimethylamino)crotylamides and -enones, propargylamides, sulfonyl fluorides, arenesulfonates, haloacetamides, cyanamides, and azine-derived nitriles, as well as electrophiles more specific for lysine residue, includingsuccinimides, thioesters, salicylic aldehydes, o-carbonylboronic derivatives, and saccharine (benzo[d]isothiazole 1,1-dioxide) derivatives were used in substructure search for the compounds to be included in the library.
Further selection process included filtering by physico-chemical parameters according to extended Ro3, removal of trivial chemotypes and visual inspection.
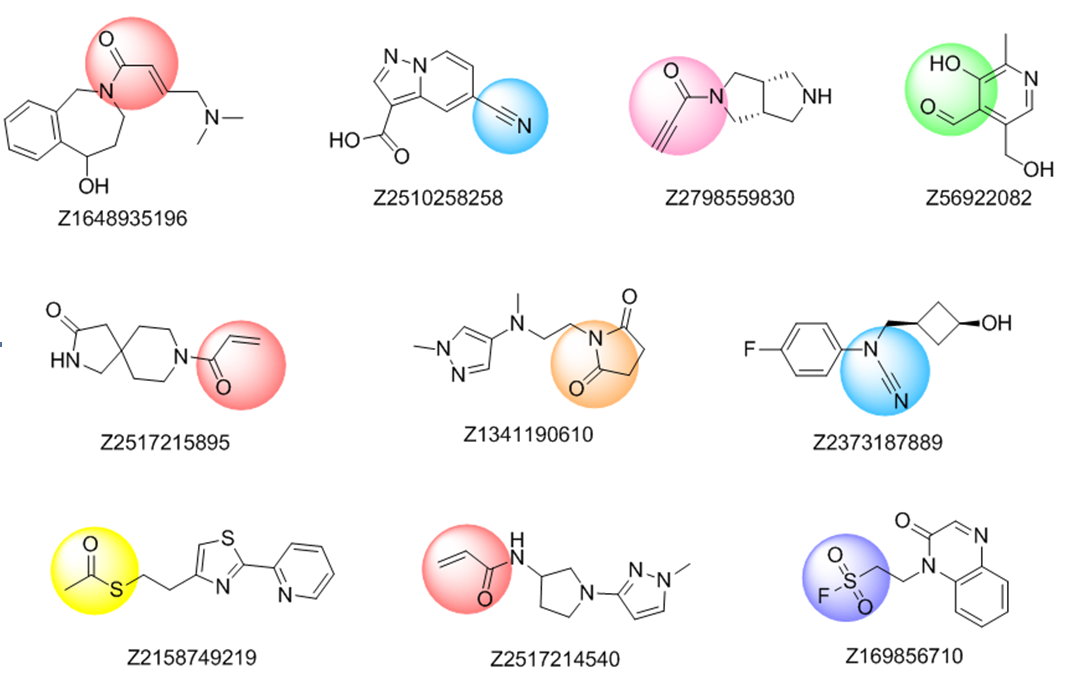
Representative examples from Lysine-focused covalent fragments library
Please contact us here to request a quote for this library or find out more about other available Enamine fragment libraries.