Tubulin Library
Library of potential tubulins ligands
3 200 compounds
Tubulin targeted library was designed using a combination of different approaches, including molecular docking, substructure and similarity, topological analogs search, molecular parameters restrictions, and specially developed structural filters.
Tubulin library is available in pre-plated format and can be quickly delivered in any customized format.
Typical Formats
Catalog No.
TBL-3200-0-Y-10
Compounds
3 200
10 plates
Amount
Assay-ready format < 300 nL
Plates and formats
384-well plates, 320 cmpds per plate, first two and last two columns empty
Price
This email address is being protected from spambots. You need JavaScript enabled to view it.
Catalog No.
TBL-3200-10-Y-10
Compounds
3 200
10 plates
Amount
10 µL of 10mM
DMSO solutions
Plates and formats
384-well plates, 320 cmpds per plate, first two and last two columns empty
Price
This email address is being protected from spambots. You need JavaScript enabled to view it.
Catalog No.
TBL-3200-50-Y-10
Compounds
3 200
10 plates
Amount
50 µL of 10mM
DMSO solutions
Plates and formats
384-well plates, 320 cmpds per plate, first two and last two columns empty
Price
This email address is being protected from spambots. You need JavaScript enabled to view it.
Download SD files
Library design
Protein structures recorded recently in PDB were considered and analyzed to be most suitable for in silico screening: 4YJ2, 5C8Y, 5CA1. The main features of protein-ligand interaction in the binding site of all three tubulin structures are very similar and could be superimposed. Hence, the protein docking model was built based on the features of critical amino acid residues in the binding pocket and ligand interactions observed in analyzed protein structures.
The docking models have been validated with a reference set of known actives (110 ligands) and nonactive molecules. Molecular queries and docking constraints were corrected according to the activity of the reference compound set.
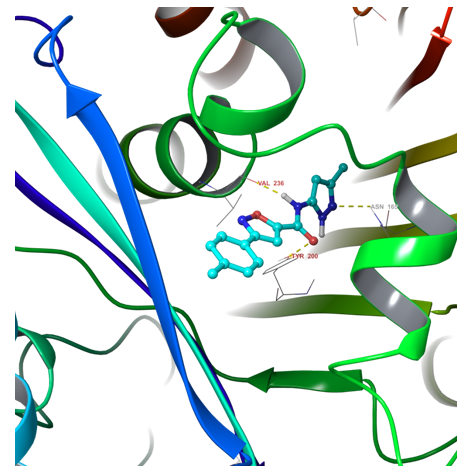
Fig. 1. Example of hit binding confirmation after docking calculation
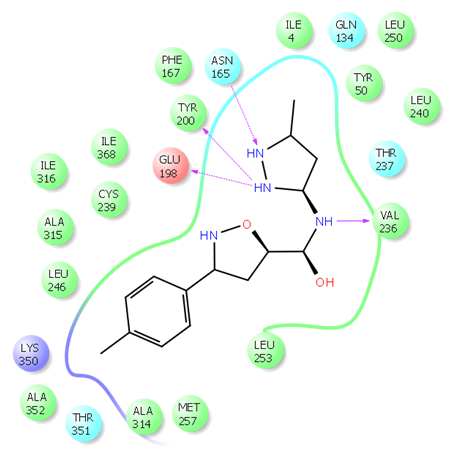
Fig. 2. 2D ligand interaction diagram of hit compound Z666232790 with a high docking score
2D Similarity search & topological analogues search
- The reference compound set was carefully compiled from available literature sources and databases: ChEMBL, BindingDB, and PudChem.
- A Tanimoto similarity range of 95–80% was used for compound selection.
- Topological fields and bioisosteric group replacement were used to search for analogs of the most potent tubulin inhibitors.
Examples of active molecules
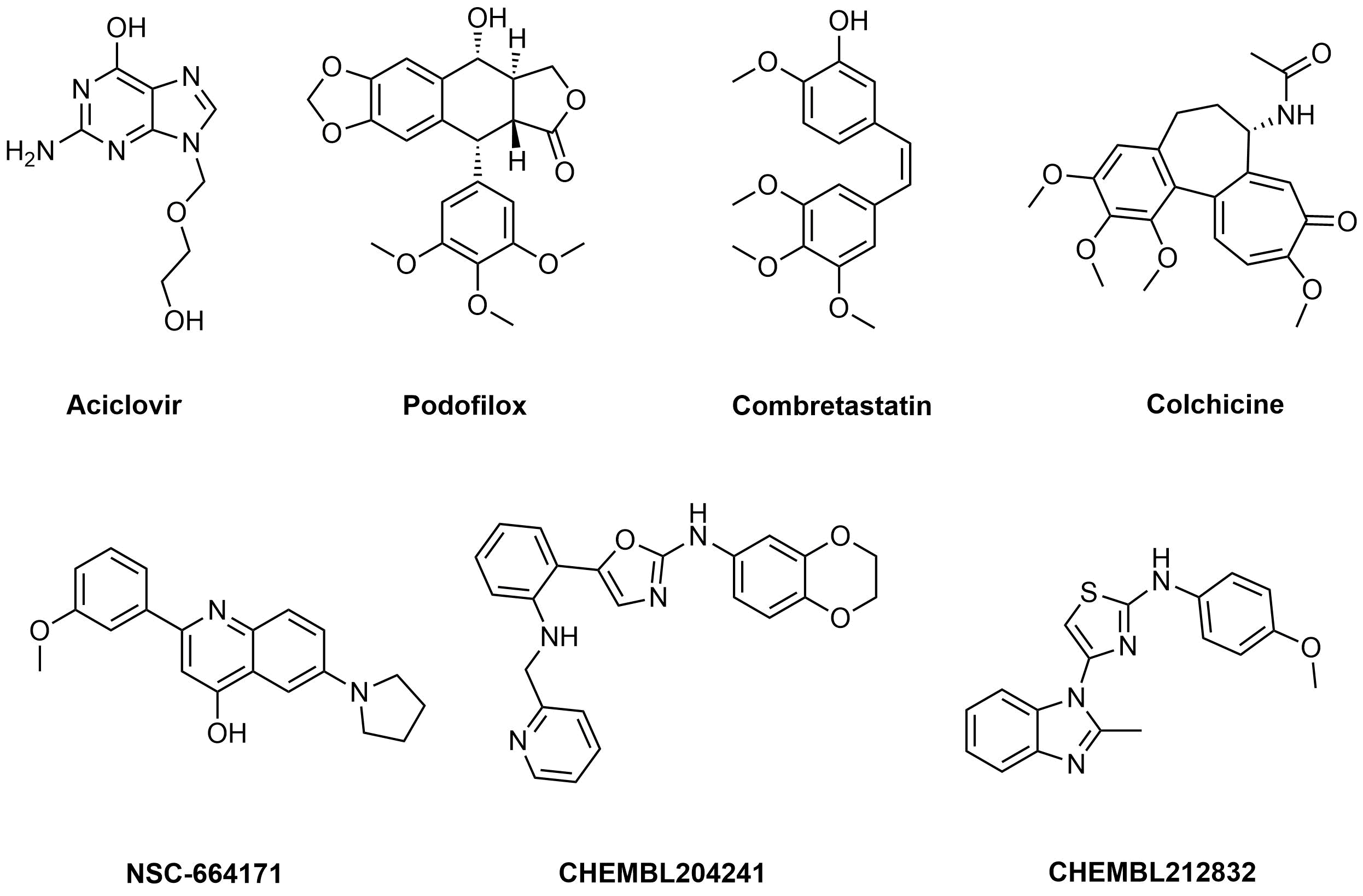
Approximately 900 compounds were selected using this approach.
Examples of molecules in Tubulin Targeted Library
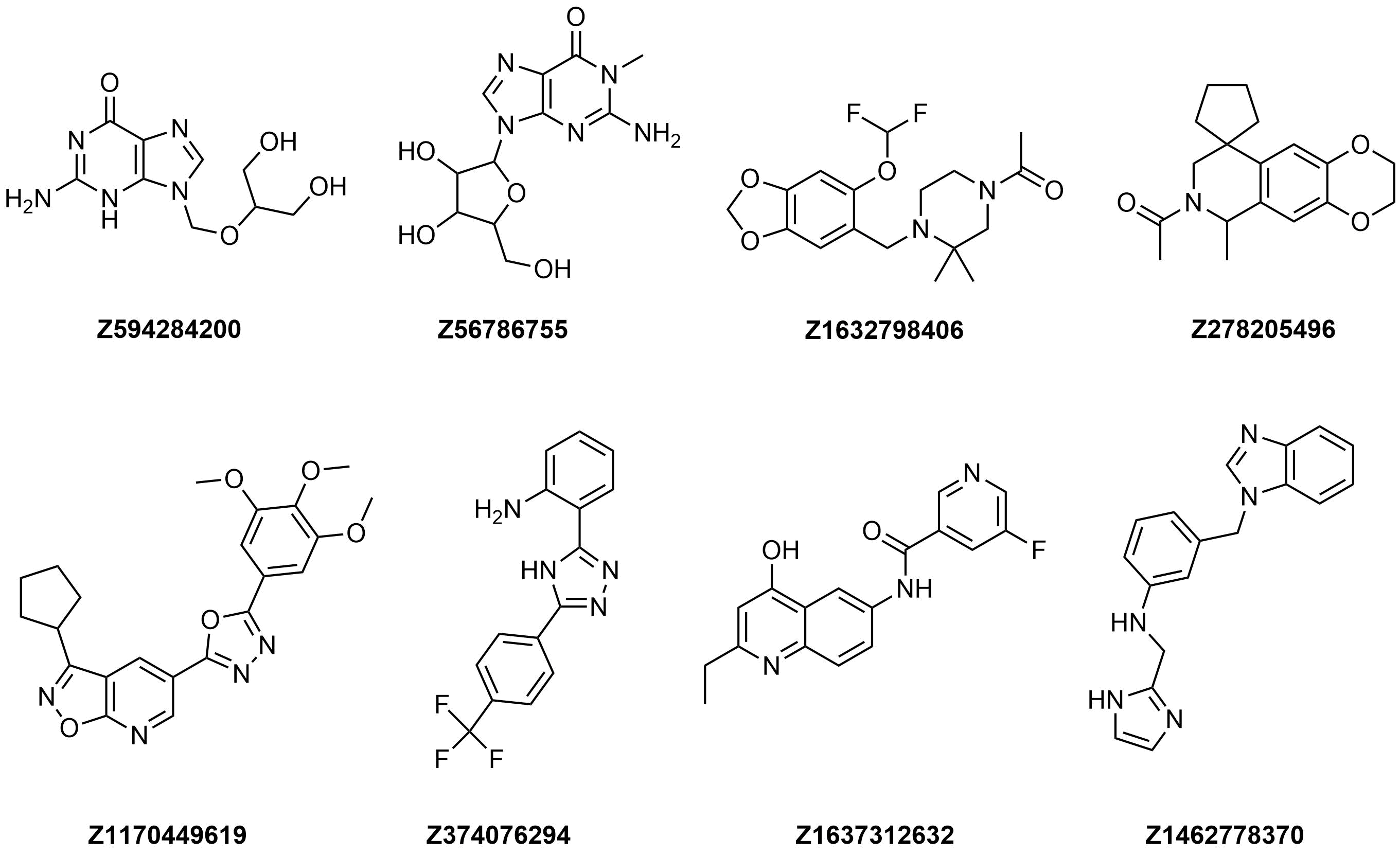
Filters applied to the Library
- In-house developed MedChem filters of unwanted structural fragments were applied to Enamine stock compound Collection as a pre-selection procedure.
- PAINS, Eli Lilly, REOS, and trivial functionalities filters included.
- Full Rule of Five compliance.


